La complexité moléculaire apprivoisée

1. Reaction Cascades
There is an undeniable interest from organic chemists and from the pharmaceutical industry to access complex alkaloids in short order. It is well established that one-pot reaction cascades or sequences are an excellent avenue to increase molecular complexity in a single operation. However, there are enormous challenges associated with this strategy: because all required functional groups must be present on the substrate for the key transformation, we have to address issues of chemoselectivity, diastereoselectivity, and regioselectivity. Over the past years, we generated the skeletons of a wide span of alkaloids of biological interest by successfully developing several reaction cascades such as those detailed below.
Sequence of Vislmeier-Haack Cyclization and Intramolecular Azomethine Ylide Cycloaddition
Daphnyphyllum Alkaloids
This one-step cyclization sequence is the most impressive we have developed. On highly functionalized linear substrates, we can activate the amide with perfect chemoselectivity and then generate a first cycle by nucleophilic trapping. Subsequent generation of an azomethine ylide in situ finally allows intramolecular cycloaddition. We then applied this approach to the construction of the tetracyclic backbone of two of the 14 Daphnyphyllum alkaloid subfamilies known for more than 35 years but for which no synthesis has been completed to date. Our approach allowed the construction of the most advanced compound published so far in these 2 subfamilies, by generating 3 cycles and 5 stereogenic centers in one operation. effectiveness of a cascading cyclization approach.These applications clearly establish the efficiency and high potential of this new reaction cascade, which will tremendously facilitate the construction of polycyclic natural products.
Lévesque, F.; Bélanger,* G. «A Versatile Cascade of Intramolecular Vilsmeier-Haack – Azomethine Ylide Generation – 1,3-Dipolar Cycloaddition Toward Tricyclic Cores of Alkaloids» Org. Lett. 2008, 10, 4939-4942
Bélanger,* G.; Boudreault, J.; Lévesque, F. «Synthesis of the Tetracyclic Core of Daphnilactone B-Type and Yuzurimine-Type Alkaloids» Org. Lett. 2011, 13, 6204-6207
Boudreault, J.; Lévesque, F.; Bélanger,* G. «Studies toward Total Synthesis of (+/-)-Caldaphnidine C via One-Pot Sequential Intramolecular Vilsmeier-Haack and Azomethine Ylide 1,3-Dipolar Cycloaddition» J. Org. Chem. 2016, 81, 9247-9268
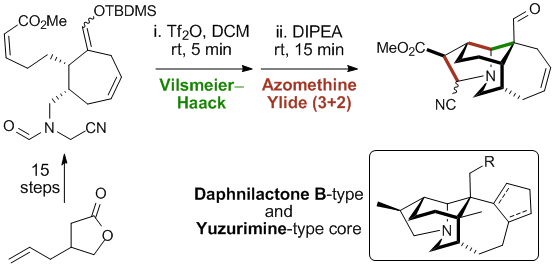
Calyciphylline B - Type Alkaloids
A suitably functionalized tricyclic adduct containing the common A,B,E rings found in calyciphylline B–type alkaloids was obtained in 9 linear steps. The key transformation features an efficient one-pot sequence of intramolecular Vilsmeier-Haack cyclization and azomethine ylide 1,3-dipolar cycloaddition in which three cycles, three new carbon-carbon bonds and three stereocenters are formed, one being fully substituted. This work also demonstrates the first use of a chiral, non racemic cyclic enol ether as an internal carbon nucleophile.
Boissarie, P.; Bélanger,* G. «Short Approach toward the Nonracemic A,B,E Tricyclic Core of Calyciphylline B-Type Alkaloids» Org. Lett. 2017, 14, 3739-3742
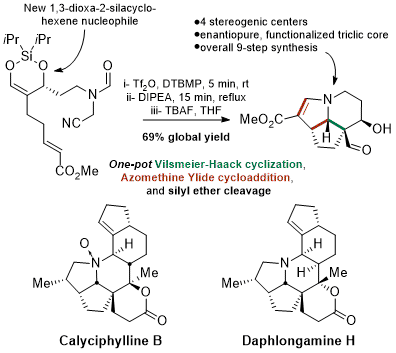
Aspidospermatan Alkaloids
The development of a new one-pot reaction sequence afforded the tricyclic core of several aspidospermatan-type alkaloids from a linear, densely functionalized substrate. The key sequence features a highly chemoselective formamide activation that triggered a Vilsmeier-Haack cyclization, followed by an azomethine ylide generation and intramolecular cycloaddition. The choice of nucleophile, azomethine ylide precursor and dipolarophile was crucial to the success of the sequence.
Hauduc, C.; Bélanger,* G. «General Approach toward Aspidospermatan-Type Alkaloids Using One-Pot Vilsmeier-Haack Cyclization and Azomethine Ylide Cycloaddition» J. Org. Chem. 2017, 82, 4703-4712
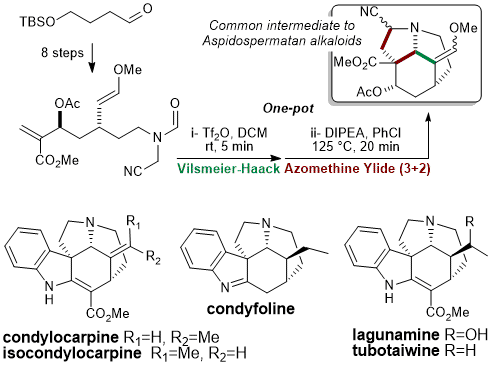
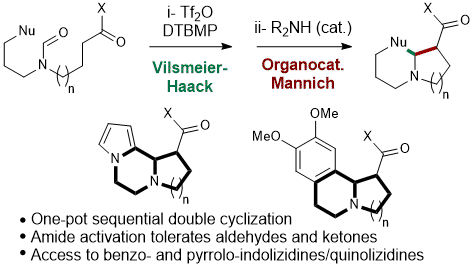
Cascade of Vilsmeier-Haack and (Organocatalyzed) Mannich Cyclizations
Indolizidines and quinolizidines
Substituted indolizidines and quinolizidines are attractive molecular scaffold because of their high occurrence in natural products exhibiting notable biological activity, as represented by the Erythrina family (erythraline shown), (+)-oleracein E, (+)-harmicine, (–)-tangutorine, and vincamine. These scaffolds could also be found in several drugs developed by the pharmaceutical industry.
Vilsmeier-Haack and Mannich Cyclization Cascade
A new approach to the synthesis of quinolizidines involving a cascade of nucleophilic cyclizations triggered by chemoselective amide activation is reported. Particular attention was given to the effect of the nature of the tethered nucleophiles on the cascade of cyclizations. As a result, simple acyclic amides gave rapid access to functionalized quinolizidines bearing either a tertiary or quaternary center at the ring junction.
Bélanger,* G.; O’Brien, G.; Larouche-Gauthier, R. «Rapid Assembly of Quinolizidines via Consecutive Nucleophilic Cyclizations onto Activated Amides» Org. Lett. 2011, 13, 4268-4271
Bélanger,* G.; Larouche-Gauthier, R.; Ménard, F.; Nantel, M.; Barabé, F. «Intramolecular Additions of Various p-Nucleophiles to Chemoselectively Activated Amides and Application to the Synthesis of Tashiromine» J. Org. Chem. 2006, 71, 704-712
Bélanger,* G.; Larouche-Gauthier, R.; Ménard, F.; Nantel, M.; Barabé, F. «Addition of Tethered Non-Aromatic Carbon Nucleophiles to Chemoselectively Activated Amides» Org. Lett. 2005, 7, 4431-4434
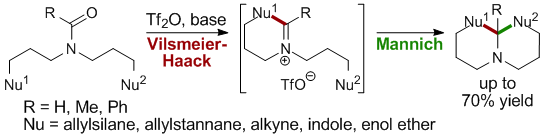
Total Synthesis of Virosine A
The development of a new one-pot sequential cyclizations involving a Vilsmeier-Haack reaction followed by an organocatalyzed Mannich reaction proved to be a valuable synthetic strategy to give access to functionalized indolizidines and quinolizidines in one operation from readily synthesized precursors.
Org. Lett. 2008, 10, 4501-4504
J. Org. Chem. 2012, 77, 3215-3221
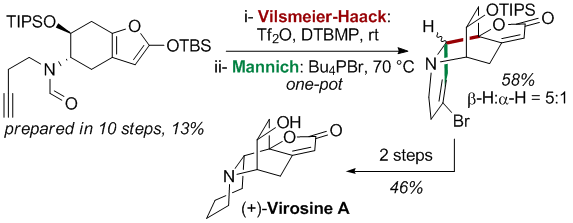
Vilsmeier-Haack and Organocatalyzed Mannich Cyclization Cascade
The methodology was further expanded to an organocatalyzed version of the Mannich reaction as the second cyclization step. This improved the span of nucleophiles that could be used and proved to be a valuable synthetic strategy to give access to functionalized indolizidines and quinolizidines in one operation from readily synthesized precursors.
J. Org. Chem. 2020, 85, 4712-4729
Synthesis of Highly Substituted β-Amino Acids
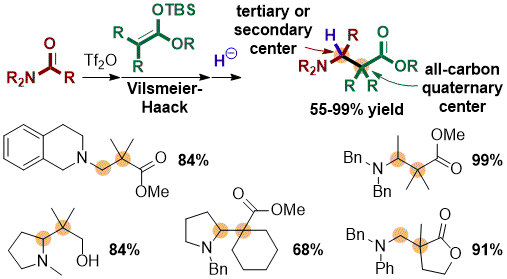
A good proportion of candidate molecules for drug development or as molecular probes for imaging are peptide-based, which causes significant problems often due to low stability against enzymatic degradation. For decades, chemists have worked to develop solutions to this problem by substituting natural amino acids by amide isosteres (e.g. fluoroalkene), non-natural α-amino acids, and β-amino acids. The latter offer a significant advantage over the first two: by the addition of a carbon atom between the acid and the amine function, it is possible to increase the degree of substitution (and targeted functionalization) on β-amino acids to a level that could not be reached with amide isosteres or β-amino acids. Another benefit often overlooked is the ability to predict and induce local secondary structure (β-sheet, alpha-helix) based on the substitution pattern of β-amino acids. Access to highly substituted β-amino acids is therefore of crucial importance for all applications involving peptides as molecular recognition units. However, the production of these molecules is still quite challenging nowadays. We recently published a novel, convergent, and versatile access to β2,2- and β2,2,3-amino acids, bearing a quaternary center adjacent to a secondary or tertiary center, respectively, based on our amide activation method and Vilsmeier-Haack trapping with ketene silyl acetals. Such highly substituted β-amino acid derivatives are essentially not accessible using previously reported methods.
Org. Lett. 2015, 17, 322-325
2. Medicinal Chemistry
Design and Synthesis of New Agonists of the Keap1/Nrf2/ARE Pathway
